

Клеточный гомеостаз в организме здорового человека определяется балансом между гибелью и пролиферацией клеток. Дефекты, возникающие в процессах дефференцировки и новообразования клеток, ведут к самоуничтожению этих структур [2]. Может показаться парадоксальным, что стимуляция клеточной гибели может способствовать выживанию организма.
Механизм, отвечающий за инициирование и выполнение запрограммированной гибели клеток, называется апоптозом. Он осуществляется под действием внеклеточных или внутриклеточных факторов. Под воздействием этого процесса, ДНК распадается на фрагменты, клетка сжимается, клеточные мембраны разрушаются, происходит элиминация и она поглощается соседней клеткой или специфичной клеткой имунной системы. Особенностью этого процесса является то, что мембрана клетки не разрушается до полного завершения этапов самопроизвольной гибели. Что дает возможность избежать риска возникновения воспалительных процессов. Обычно от начала запуска апоптоза до окончательной клеточной фрагментации требуется несколько часов. Однако этот период зависит от типа клетки, стимула и апоптотического пути [1,2].
Апоптозные клетки выглядят как округлые либо овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина [1]. (Рис. 1.).
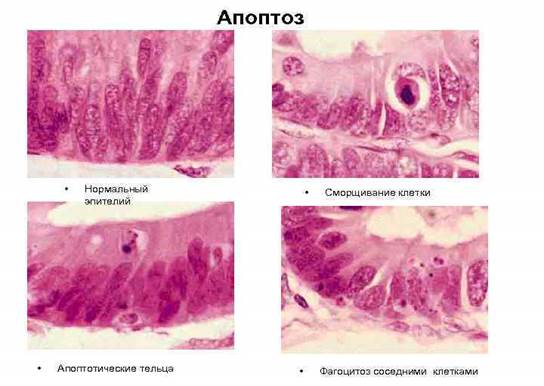
Рис. 1. Стадии апоптоза эпителиальной клетки
Основными регуляторами запрограммированной гибели клеток являются белки, принадлежащие к семейству Bcl-2. Эти белки могут, как активировать апоптоз, то есть быть проапоптотическими, так и ингибировать его, обладая антиапоптотическими свойствами. Антиапоптотические белки в здоровой клетке связывают и инактивируют проапоптотические. Это происходит тогда, когда она не нуждается в гибели [4,5].
Существует два основных пути апоптоза: внутренний и внешний. Внешний путь обеспечивает связывание «лиганд смерти» с «рецептором смерти». Внутренний путь контролируется митохондриями и выделением цитохрома С. Регуляторы апоптоза взаимодействуют со специфическими рецепторами на мембране клетки, называющимися «рецепторы смерти», которые связываются с молекулами, сигнализирующими гибель, как часть внешнего апоптотического пути. Это связывание вызывает эффект апоптоза [7].
Контроль такой элиминации производится в митохондриях, которые обеспечивают внутреннюю часть апоптического пути. В этих органеллах содержатся сигнальные молекулы, связаные с митохондриальной мембраной и известные как цитохром C. В ответ на проапоптотические сигналы из проапоптотических белков, высокую концентрацию Ca 2+ в цитозоле или гипоксию цитохром C высвобождается в клетку митохондриями и связывается с белком. Это приводит к образованию апоптосомы. После образования апоптосома активирует группу белков под названием каспазы, которые денатурируют другие белки в клетках [8]. Так как активные каспазы могут разрушающе воздействовать внутри здоровой клетки, они производятся в неактивной форме – прокаспазы. В фазе апоптоза семейство каспаз представляет собой основные эффекторные молекулы самого процесса элиминации, которые вносят вклад в конечные стадии апоптотической гибели клеток путем компонентов цитоскелетного аппарата и ядерной ДНК [4,9]. Все каспазы подразделяются на инициаторы, эффекторы и стимуляторы. Инициаторы расщепляют и активируют каспазы эффекторы, амплифицируя сигнал. Эффекторы расщепляют различные белки, что приводит к процессу апоптоза. Активация каспаз ведет к запуску протеолитического каскада реакций, провоцирующих гибель клетки [9].
Помимо каспаз, чрезвычайно важным является белок P53, обеспечивая обнаружение повреждения ДНК, аномалий хромосом и остановку клеточного цикла. Если повреждения необратимые, то апоптоз актимируется. P53 активирует процесс путем увеличения продуцирования проапоптотического белка, который активирует каспазный каскад, что в конечном итоге приводит к самоуничтожению клетки [10].
Апоптоз — это защита организма от персистенции пораженных клеток, которые могут оказаться потенциально опасными для многоклеточного организма. Однако, эти процессы могут нарушаться, сопровождаясь либо чрезмерным апоптозом, например, в случае дегенеративных заболеваний, либо недостаточным, что приводит к ускоренной пролиферации дефектных клеток и возникновению онкологии [2]. Проблемы недостаточного самоуничтожения клеток могут возникать на любом этапе апоптоза, что приводит к злокачественной трансформации пораженных клеток, метастазированию опухолей и устойчивости к противоопухолевым препаратам [6]. Следовательно, устойчивость к апоптозу или угнетение этого процесса играют жизненно важную роль в канцерогенезе. Одной из причин торможения процессов самопроизвольной гибели клеток является нарушения баланса проапоптотических и антиапоптотических белков. Проапоптотические (например, Bax, Bad, Bcl-Xs) и антиапоптотические белки (например, Bcl-2, Bcl-XL, Mcl-L и т. Д.) являются двумя основными группами белков семейства Bcl -2. Антиапоптотические белки регулируют апоптоз, блокируя выделение цитохрома С в митохондриях, в то время как проапоптические -стимулируют . Нарушение этого баланса вызывает угнетение процесса апоптоза в пораженных клетках. Причиной может стать как чрезмерная экспрессия антиапоптотических белков, так и проапоптотических или их комбинации.
Так, в основе возникновения В-клеточной лимфомы лежит механизм, подавляющий синтез проапоптического белка семейства Bcl-2, что приводит к торможению апоптоза клеток фолликулярной лимфомы и их пролиферацию [3]. (Рис. 2.).
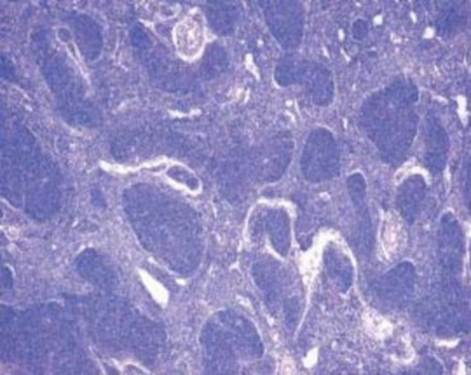
Рис. 2. Гистопрепарат ткани лимфатического узла. Фолликулярная лимфома.
Новообразования можно рассматривать как результат последовательности генетических изменений, в течение которых нормальная клетка превращается в злокачественную. И именно несостоявшийся апоптоз таких клеток является одним из существенных критериев, которые вызывают злокачественную трансформацию [3]. Злокачественные клетки подвергаются серии генетических изменений. Если это способствует их преимущественному росту над нормальными клетками, то риск развития и роста новообразований значительно возрастает. Например, когда в клетках кожи возникают повреждения под воздействием ультрафиолетового излучения (например, солнцем, соляриями), обычно срабатывает апоптоз. Это помогает устранить патологические элементы. Если апоптоз не происходит, такие клетки могут выживать и пролифирировать, превращаясь в злокачественные. (Рис.3.).
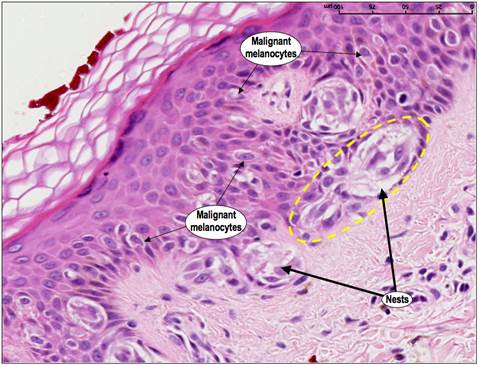
Рис. 3. Меланома.
Запрограммированная гибель также играет роль в распространении онкологического процесса. Чтобы злокачественная клетка переместилась в другую часть тела ( метастазирование), она должна уметь выживать в крови или лимфатических системах и проникать в окружающие ткани. Апоптоз предотвращает эти процессы [6]. Наиболее распространенным механизмом уклонения от гибели является потеря предшественника апоптоза - белка P53 или его мутация. Более половины всех онкологических пациентов имеют мутированный или отсутствующий ген, программирующий синтез р53, что приводит к повреждению или отсутствию белка. При некоторых опухолевых процессах р53 трансформируется из активатора апоптоза в ингибитор, тем самым он не только не индуцирует апоптоз, а его блокирует. Это же провоцирует метастазирование новообразований, так как позволяет обходиться злокачественным клеткам без специфического микроокружения, избегая самопроизвольной гибели [3,9]. Однако, ингибирование процессов апоптоза не единственное условие для роста и развития новообразований. Без устойчивого ангиогенеза, самодостаточности сигналов роста и нечувствительности к сигналам против роста, «прото-раковые» клетки все еще могут умирать другими способами, даже если они избегают апоптотической запрограммированной клеточной смерти [6].
Выводы. Таким образом, канцерогенез можно рассматривать как сложный клеточный процесс, который связан с неограниченным репликативным потенциалом, независимостью от сигналов роста и параллельным сопротивлением ингибирующему рост сигналу, уклонение от активации клеточной смерти, устойчивый ангиогенез, а также способность тканевой инвазии и метастазирования. Злокачественные опухоли являются инвазивными и могут метастазировать в отдаленные места через систему кровообращения. Следовательно, метастатическое распространение, а не первичная опухолевая нагрузка, является основной причиной смертей от рака [6,4,7].
Библиографическая ссылка
Кутузова Л.А., Тончева К.С. РОЛЬ АПОПТОЗА В КАНЦЕРОГЕНЕЗЕ // Международный студенческий научный вестник. – 2018. – № 6. ;URL: https://eduherald.ru/ru/article/view?id=19316 (дата обращения: 24.04.2024).